WDL (pronounced widdle) is a workflow description language to define tasks and workflows. WDL aims to describe tasks with abstract commands that have inputs, and once defined, allows you to wire them together to form complex workflows.
Learn More
CROMWELL is the execution engine (written in Java) that supports running WDL scripts on three types of platforms: local machine (e.g. your laptop), a local cluster/compute farm accessed via a job scheduler (e.g. Slurm, GridEngine) or a cloud platform (e.g. Google Cloud or Amazon AWS).
Learn More
Introduction
Pre-requisites: This tutorial assumes that you have an understanding of the basic structure of a WDL script. Learn here
This tutorial will walk you through steps for creating a WDL script, and executing it on the HPC system. For the purpose of this document, we will write a (very) basic real-world workflow that does something useful!
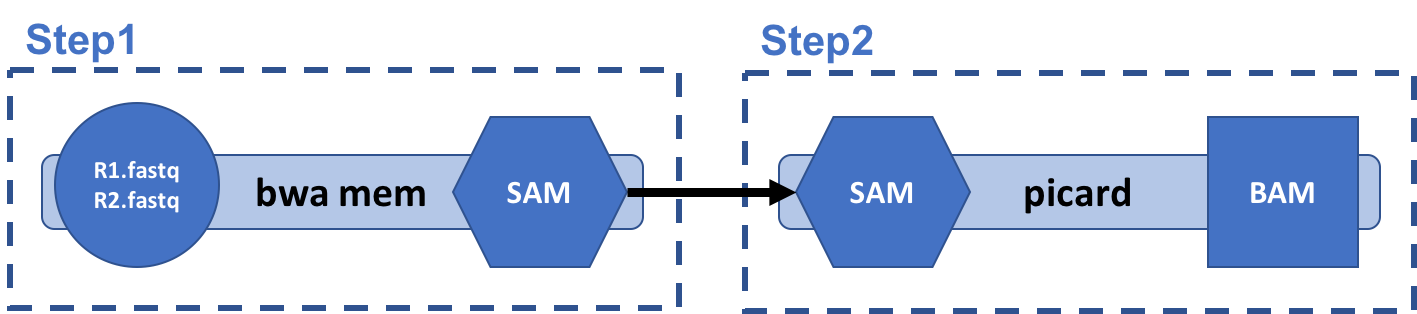
Our workflow:
The processing with bwa-mem contains two tasks:
- Alignment of sequence files to reference genome using bwa, followed by
- SAM to BAM format conversion using
picard.
The tasks are joined together using linear chaining, with output from bwa step used as input to the picard step.
Inputs:
- Sample paired-end FASTQ files
- hg38 reference fasta and BWA index files
UVA HPC modules:
Set up your Working Environment
- Login to UVA HPC and create a root working directory in your
/scratch folder:
cd /scratch/$USER/
mkdir wdl_tutorial
cd wdl_tutorial
-
Get the Sample FASTQ files. Copy the sample paired-end fastq files to this folder. For this tutorial, we will use reads for NA12878, downloaded from here. You can download the dataset, or use DNA-seq data for any sample of your choice.
-
Get the reference genome files. We will use the hg38 reference fasta and BWA indexes from the genomes repo on the HPC system at /project/genomes/Homo_sapiens/UCSC/hg38/Sequence/BWAIndex/. All UVA HPC users have read access to these reference genomes, no need to download/copy them to the working directory!
WDL script
First, let’s write our workflow! Open a blank text file in your favorite text editor and save it as bwaAln.wdl.
Workflow
Let’s begin with the workflow skeleton. Our workflow – let’s name it bwa_mem – calls two tasks: align and samSort.
workflow bwa_mem {
call align { input: }
call samSort { input: }
}
Next, we need to tell cromwell how to link the tasks together. We will tell samSort task to take the outsam from align task as its input. So let’s update the workflow definition:
workflow bwa_mem {
call align { input: }
call samSort {
input:
insam = align.outsam
}
}
Finally, let’s add the inputs for our first task, as well as few variables each task is going to need.
workflow bwa_mem {
String sample_name
File r1fastq
File r2fastq
File ref_fasta
File ref_fasta_amb
File ref_fasta_sa
File ref_fasta_bwt
File ref_fasta_ann
File ref_fasta_pac
call align {
input:
sample_name = sample_name,
r1fastq = r1fastq,
r2fastq = r2fastq,
ref_fasta = ref_fasta,
ref_fasta_amb = ref_fasta_amb,
ref_fasta_sa = ref_fasta_sa,
ref_fasta_bwt = ref_fasta_bwt,
ref_fasta_ann = ref_fasta_ann,
ref_fasta_pac = ref_fasta_pac
}
call sortSam {
input:
sample_name = sample_name,
insam = align.outsam
}
}
Our workflow definition in bwaAln.wdl is complete!
Tasks
1. align
This task will align the paired-end reads to hg38 build of human reference genome, using bwa mem algorithm. Here’s the skeleton definition:
task align {
Inputs/Variables
command {...}
runtime {...}
output {...}
}
Now let’s define the alignment command and the variables we need to execute it:
task align {
String sample_name
File r1fastq
File r2fastq
File ref_fasta
File ref_fasta_amb
File ref_fasta_sa
File ref_fasta_bwt
File ref_fasta_ann
File ref_fasta_pac
Int threads
command {
bwa mem -M -t ${threads} ${ref_fasta} ${r1fastq} ${r2fastq} > ${sample_name}.hg38-bwamem.sam
}
runtime {...}
output {...}
We are passing the paired-end fastq files for our sample, the reference fasta and its BWA indexes, as well as the number of threads for alignment as inputs to the task. Notice how to reference the variables in the command, using ${variable_name}.
Next, add runtime attributes, i.e. the number of cpus (same as num of threads passed as variable) and memory (16GB) required for the task.
...
runtime {
cpus: threads
requested_memory_mb: 16000
}
...
Finally, lets define out output:
...
output {
File outsam = "${sample_name}.hg38-bwamem.sam"
}
...
Our task is complete and should look like this:
task align {
String sample_name
File r1fastq
File r2fastq
File ref_fasta
File ref_fasta_amb
File ref_fasta_sa
File ref_fasta_bwt
File ref_fasta_ann
File ref_fasta_pac
Int threads
command {
bwa mem -M -t ${threads} ${ref_fasta} ${r1fastq} ${r2fastq} > ${sample_name}.hg38-bwamem.sam
}
runtime {
cpus: threads
requested_memory_mb: 16000
}
output {
File = "${sample_name}.hg38-bwamem.sam"
}
}
**2. sortSam**
This task will take the output from alignment step in SAM format, convert it to BAM, sort it on coordinates and create the index using picard SortSam utility. This the task skeleton:
task sortSam {
Inputs/Variables
command {...}
runtime {...}
output {...}
}
Add variables …
task sortSam {
String sample_name
File insam
...
}
Add command …
...
command <<<
java -jar $EBROOTPICARD/picard.jar \
SortSam \
I=${insam} \
O=${sample_name}.hg38-bwamem.sorted.bam \
SORT_ORDER=coordinate \
CREATE_INDEX=true
>>>
...
}
Note: picard is available as a module on the HPC system. When you load the module to your environment (using module load picard), it also defines the $EBROOTPICARD environment variable, which defines the full path to the jar file for calling picard utilities.
Add output …
...
output {
File outbam = "${sample_name}.hg38-bwamem.sorted.bam"
File outbamidx = "${sample_name}.hg38-bwamem.sorted.bai"
}
....
For this task, we don’t need custom runtime attributes, and will be using the default described in the backend configuration file for UVA HPC!
The complete sortSam task definition should look like this:
task sortSam {
String sample_name
File insam
command <<<
java -jar $EBROOTPICARD/picard.jar \
SortSam \
I=${insam} \
O=${sample_name}.hg38-bwamem.sorted.bam \
SORT_ORDER=coordinate \
CREATE_INDEX=true
>>>
output {
File outbam = "${sample_name}.hg38-bwamem.sorted.bam"
File outbamidx = "${sample_name}.hg38-bwamem.sorted.bai"
}
}
Complete WDL script - bwaAln.wdl
##
## My First WDL/CROM workflow on Rivanna
##
## Description:
## This WDL workflow will align paired-end sequences of a sample to
## hg38 build of human genome using bwa mem algorithm, followed by
## sorting and indexing the alignment map using picard
##
## This workflow is designed for demonstration purpose only!
############
# Workflow #
############
workflow bwa_mem {
String sample_name
File r1fastq
File r2fastq
File ref_fasta
File ref_fasta_amb
File ref_fasta_sa
File ref_fasta_bwt
File ref_fasta_ann
File ref_fasta_pac
call align {
input:
sample_name = sample_name,
r1fastq = r1fastq,
r2fastq = r2fastq,
ref_fasta = ref_fasta,
ref_fasta_amb = ref_fasta_amb,
ref_fasta_sa = ref_fasta_sa,
ref_fasta_bwt = ref_fasta_bwt,
ref_fasta_ann = ref_fasta_ann,
ref_fasta_pac = ref_fasta_pac
}
call sortSam {
input:
sample_name = sample_name,
insam = align.outsam
}
}
#########
# Tasks #
#########
## 1. This task will align the reads to reference
## using bwa mem algorithm
task align {
String sample_name
File r1fastq
File r2fastq
File ref_fasta
File ref_fasta_amb
File ref_fasta_sa
File ref_fasta_bwt
File ref_fasta_ann
File ref_fasta_pac
Int threads
command {
bwa mem -M -t ${threads} ${ref_fasta} ${r1fastq} ${r2fastq} > ${sample_name}.hg38-bwamem.sam
}
runtime {
cpu: threads
requested_memory_mb: 16000
}
output {
File outsam = "${sample_name}.hg38-bwamem.sam"
}
}
## 2. This task will sort sam by coordinate,
## convert it to BAM, and index the BAM
task sortSam{
String sample_name
File insam
command <<<
java -jar $EBROOTPICARD/picard.jar \
SortSam \
I=${insam} \
O=${sample_name}.hg38-bwamem.sorted.bam \
SORT_ORDER=coordinate \
CREATE_INDEX=true
>>>
output {
File outbam = "${sample_name}.hg38-bwamem.sorted.bam"
File outbamidx = "${sample_name}.hg38-bwamem.sorted.bai"
}
}
Validate
Next, we will validate our script, make sure there are no syntax errors.
We will use wdltool utility toolkit that includes a syntax validation function. It is available as a module on the HPC system
module load wdltool
This will define a global environment variable, $WDLTOOLPATH, that stores the root directory path for the jar file. To validate our script, we simply call the validate function:
java -jar $WDLTOOLPATH/wdltool-0.14.jar validate bwaAln.wdl
This function parses the WDL script and alerts us to any syntax errors such missing curly braces, undefined variables, missing commas and so on. It will resolve imports, but note that it is not able to identify errors like typos in commands, specifying the wrong filename, or failing to provide required inputs to the programs that are run in the workflow.
No messages will be thrown if the syntax is valid!
Now, we will create a JSON file of inputs for our workflow. We will use the inputs function in the wdltool package to create a template to populate the values -
java -jar $WDLTOOLPATH/wdltool-0.14.jar inputs bwaAln.wdl > bwaAln.inputs.json
This will create a file called bwaAln.inputs.json that lists all the inputs to all the tasks in your script following the pattern below:
{
"<workflow name>.<task name>.<variable name>": "<variable type>"
}
This saves you from having to compile a list of all the tasks and their variables manually!
Now, populate the variables using your favorite text editor. You can verify the file’s content with the cat command:
cat bwaAln.inputs.json
Output in terminal:
{
"bwa_mem.sample_name": "NA12878",
"bwa_mem.r1fastq": "/FULL/PATH/TO/NA12878_R1.fastq",
"bwa_mem.r2fastq": "/FULL/PATH/TO/NA12878_R2.fastq",
"bwa_mem.ref_fasta": "/project/genomes/Homo_sapiens/GATK_bundle/hg38/BWAIndex/hg38.fa",
"bwa_mem.ref_fasta_amb": "/project/genomes/Homo_sapiens/GATK_bundle/hg38/BWAIndex/hg38.fa.amb",
"bwa_mem.ref_fasta_sa": "/project/genomes/Homo_sapiens/GATK_bundle/hg38/BWAIndex/hg38.fa.sa",
"bwa_mem.ref_fasta_ann": "/project/genomes/Homo_sapiens/GATK_bundle/hg38/BWAIndex/hg38.fa.ann",
"bwa_mem.ref_fasta_pac": "/project/genomes/Homo_sapiens/GATK_bundle/hg38/BWAIndex/hg38.fa.pac",
"bwa_mem.ref_fasta_bwt": "/project/genomes/Homo_sapiens/GATK_bundle/hg38/BWAIndex/hg38.fa.bwt",
"bwa_mem.align.threads": 16
}
Please provide FULL PATH to fastq files! There is no need to change file paths to hg38 reference fasta and BWA index files!
Execute
At the moment, Cromwell is the only fully-featured execution engine that supports WDL. It is available as a module on the HPC system.
module load cromwell
Basic syntax:
java -jar $CROMWELLPATH/cromwell-30.1.jar <action> <parameters>
Backend
In order to run each task of our workflow as a slurm job, we need to configure a SLRUM backend.
Create an empty text file, cromwell-rivanna.conf, and copy the contents described in this post.
Slurm batch submission script
Finally, we will write a simple Slurm submission script to execute our workflow. Create an empty file submit-bwaAln.sh and copy the below contents to it -
#!/bin/bash
#SBATCH --job-name=bwaAln # Job name
#SBATCH --cpus-per-task=16 # Number of CPU cores per task
#SBATCH --mem=16gb # Job Memory
#SBATCH --time=5:00:00 # Time limit hrs:min:sec
#SBATCH --output=bwaAln_%A.out # Standard output log
#SBATCH --error=bwaAln_%A.err # Standard error log
#SBATCH -A <YOUR_ALLOCATION # allocation groups
#SBATCH -p standard # slurm queue
pwd; hostname; date
# load modules
module load cromwell
module load bwa
module load picard
# Submit cromwell job
java -Xmx8g -Dconfig.file=~/rivanna-cromwell.conf \
-jar $CROMWELLPATH/cromwell-30.1.jar \
run bwaAln.wdl \
--inputs bwaAln.inputs.json
Note: This assumes that the rivanna-crowell.conf backend configuration file is located in your home directory. Learn more about how to create this Cromwell configuration file here.
Submit the job
submit submit-bwaAln.sh
Monitor job progress
squeue -u $USER
Outputs
The outputs of the workflow will be written to <workflow_root>/call-<call_name>/execution/ folder! Please explore the directory structure for relevant files!